一般要做弛豫计算,需要设置弛豫收敛标准,也就是告诉系统收敛达成的判据(convergence break condition),当系统检测到能量变化减小到一个确定值时例如EDIFFG=1E-3时视为收敛中断计算,移动离子位置尝试进行下一步计算。EDIFFG这个值可以为负,例如EDIFFG=-0.02,这时的收敛标准是当系统发现所有离子间作用力都小于给定的数值,如0.02eV/A时视为收敛而中断。
弛豫计算主要有两种方式:准牛顿方法(quasi-Newton RMM-DIIS)和共轭梯度法(CG)两种。准牛顿方法计算速度较快,适合于初始结构与平衡结构(势能面上全局最小值)比较接近的情况,而CG方法慢一些,找到全局最小的可能性也要大一些。选择方法为IBRION=1时为准牛顿方法而IBRION=2时为CG方法。
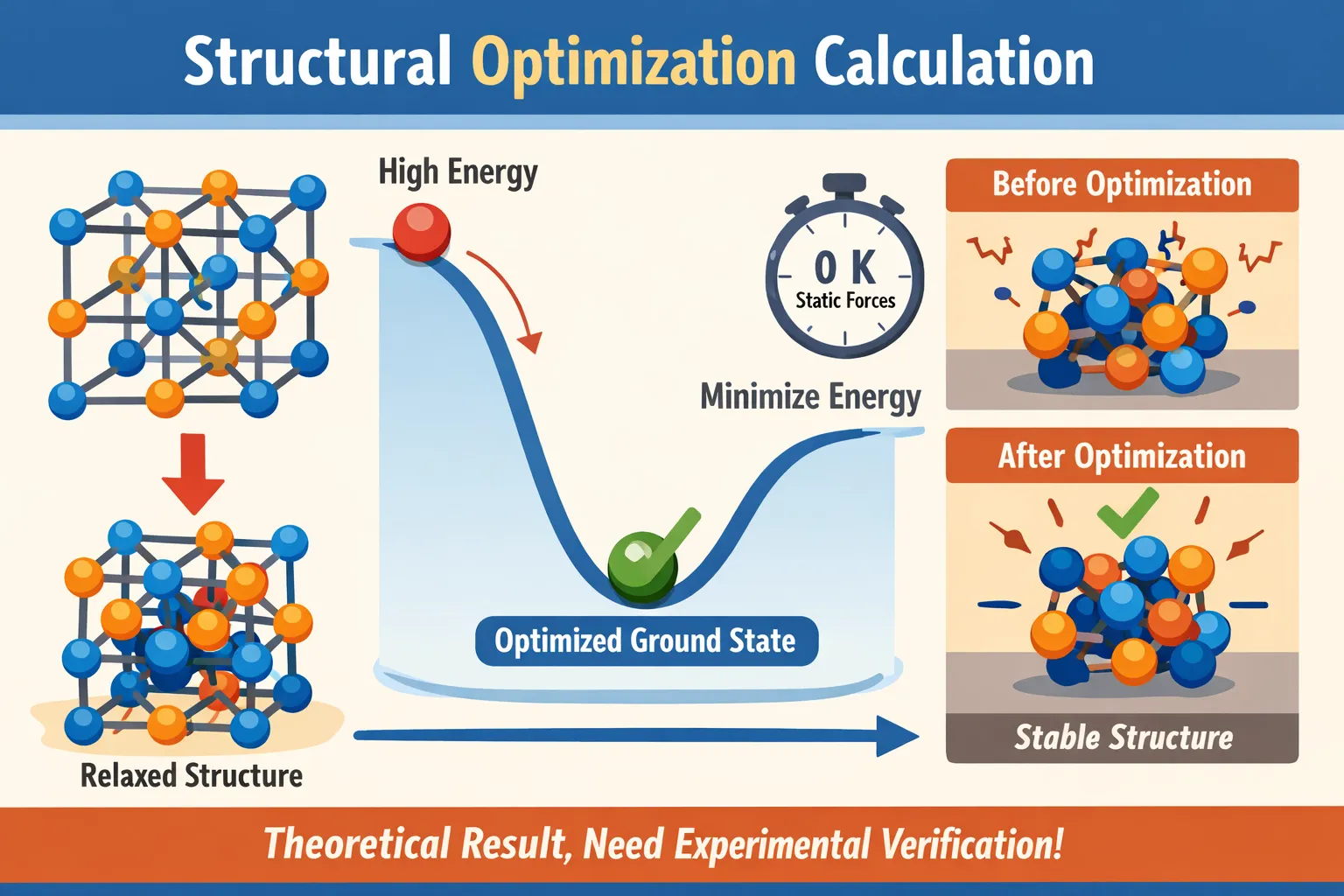
结构优化也叫结构迟豫。是指对整个输入体系的坐标进行调整,得到一个相对稳定的基态结构。结构优化分原子迟豫和电子迭代两个嵌套的过程,每次计算中都进行原子迟豫和电子迭代计算(电子迭代嵌套在原子迟豫中),达到原子迟豫收敛标准时进行下一步计算,直到达到自动中断或者最大原子迟豫步数。(通常以前后两次总自由能之差或 原子所受最大的力 作为原子迟豫的收敛标准,电子自洽迭代计算以总能作为收敛标准,默认为10-4,因电子迭代的嵌套特性,以及电子迭代和原子迟豫的收敛速度不同,最终以原子迟豫最大步数作为强制停止参数)
通过结构优化使体系达到相对稳定结构,因此可以得到体系的总能量(哪个能量??)、晶体结构参数(可以得到包括晶格常数、结合能等)
同时得到CONTCAR(离子迟豫时,每步移动后的体系的晶格参数,和POSCAR内容相同。因此结构优化完成后,要使用输出的优化CONTCAR拷贝成POSCAR进行下一步的静态自洽计算,对电子结构做进一步调整 )
导入.cif文件,用vaspkit产生POSCAR
根据.cif文件生成POSCAR
- vaspkit→1→105
关键字解释:
- SYSTEM:作业名
- ISTART:
- 0:从头开始计算
- 1:连续计算,有上一步的计算
- ICHARG:控制晶体的电荷密度
- 2:从孤立原子获得(球形电荷密度)
- 从上一步计算所获得的电荷密度
- LWAVE:控制波函数的写入
- LCHARG:控制波函数的写入
- NWRITE:控制程序输出的详细程度
- 1:普通输出
- 2:详细输出
- ENCUT:截断能,vasp计算过程中,对波函数进行平面波展开的最大动能。在进行晶体结构(三维晶体)优化,需要将参数取到ENMAX的1.3倍以上,ENMAX在赝势文件POTCAR中
- ALGO:电子自洽迭代所需算法
- F:采用Davison和RMM混合算法
- LDIAG:控制计算过程中,子空间是否对角化
- NELM:控制电子自洽迭代的最大步数
- NELMIN:控制电子自洽迭代的最小步数
- NELMDL:控制电子自洽迭代之前,进行预收敛的非自洽的步数
- EDIFF:电子自洽迭代的能量收敛精度
- ISPIN:控制自旋
- 1:一种自旋
- 2:两种自旋
- LMAXPAW:计算过程中最大角动量=2*最大角动量
- LMAXMIX:计算过程中最大角动量=2*最大角动量
- ADDGRID:电荷密度缀加。在NGXF这样的fine网格基础上,叠加第三个网格,在这个网格上进行电荷密度缀加,精细度可以达到fine网格的8倍以上
- ISMEAR:控制单电子轨道占据
- 0:高斯占据,高斯函数表示单电子轨道的占据
- SIGMA:单电子轨道热展宽,考虑一种电子熵的贡献,半导体和绝缘体取到0.1以下,金属0.1~0.2
- GGA:控制交换关联泛函
- PE:晶体结构优化常用的PBE泛函
- IVDW:范德华修正
- 11:对体系进行Grimme的第三版本的范德华修正
- IBRION:控制离子弛豫的算法
- 2:共轭梯度算法(初始结构不太好的情况,收敛比较冒进,收敛速度快)
- POTIM:控制离子弛豫的单步步长
- NSW:控制离子弛豫的最大步数
- ISIF:控制离子弛豫过程中,晶胞的应力张量是否计算
- 3:充分弛豫,即弛豫离子的位置,又弛豫晶胞的形状和体积
- 2:不计算晶胞应力张量,仅弛豫离子位置,不弛豫晶胞体积和形状
- ISYM:控制对称性
- 0:关掉对称性
- 2:对称性约束
- EDIFFG:控制离子弛豫的受力收敛精度
- -0.01:0.01eV/A
- PREC:
- A:精确的计算
pv赝势:
- 不仅仅把最外层的s电子以及d壳层的d电子作为价态来处理,把p壳层的电子作为价态来处理
输出文件
CHG和CHGCAR:电荷密度展示和计算的电荷文件
CONTCAR:结构优化获得的结构文件
DOSCAR:存放的态密度数据文件
EIGENVAL:绘制能带所需的本征值数据
IBZKPT:程序自动产生的布里渊去k点
OSIZCAR:每一个离子步和电子步的收敛信息
OUTCAR:总体的输出文件
PCDAT:对关联式文件,对于分子动力学计算有用
WAVECAR:存放体系的波函数
XDATCAR:存放结构优化的结构轨迹
step2
step1产生的CONTCAR改为POSCAR
ISIF改为2
三维晶体结构修正可以一步获得
- ISIF:3(充分弛豫)
- ISYM:2(对称性约束)
- POTIM:0.3(由于共轭梯度算法比较冒进,减小结构优化的单步步长)
| ISIF值 | 优化离子位置 | 优化晶胞形状 | 优化晶胞体积 | 计算应力 | 典型应用场景 |
|---|---|---|---|---|---|
| 2 | 是 | 否 | 否 | 否 | 固定晶胞形状和体积,仅优化原子位置。 |
| 3 | 是 | 是 | 否 | 是 | 优化晶胞形状(保持体积不变),适用于表面或界面计算。 |
| 4 | 是 | 是 | 是 | 是 | 全优化(形状+体积+离子位置),适用于体材料(如块体晶体)。 |
| 5 | 是 | 否 | 否 | 是 | 类似ISIF=2,但计算应力(不常用)。 |
| 6 | 否 | 是 | 是 | 是 | 仅优化晶胞(形状+体积),不优化原子位置(如晶格参数扫描)。 |
| 7 | 是 | 是 | 是 | 是 | 与ISIF=4相同(VASP默认值,需确认版本差异)。 |